Peer-Reviewed publications and conference papers.
2015
Ferhat Ay, William Stafford Noble. "Analysis methods for studying the 3D architecture of the genome." Genome Biology. 16:183, 2015.
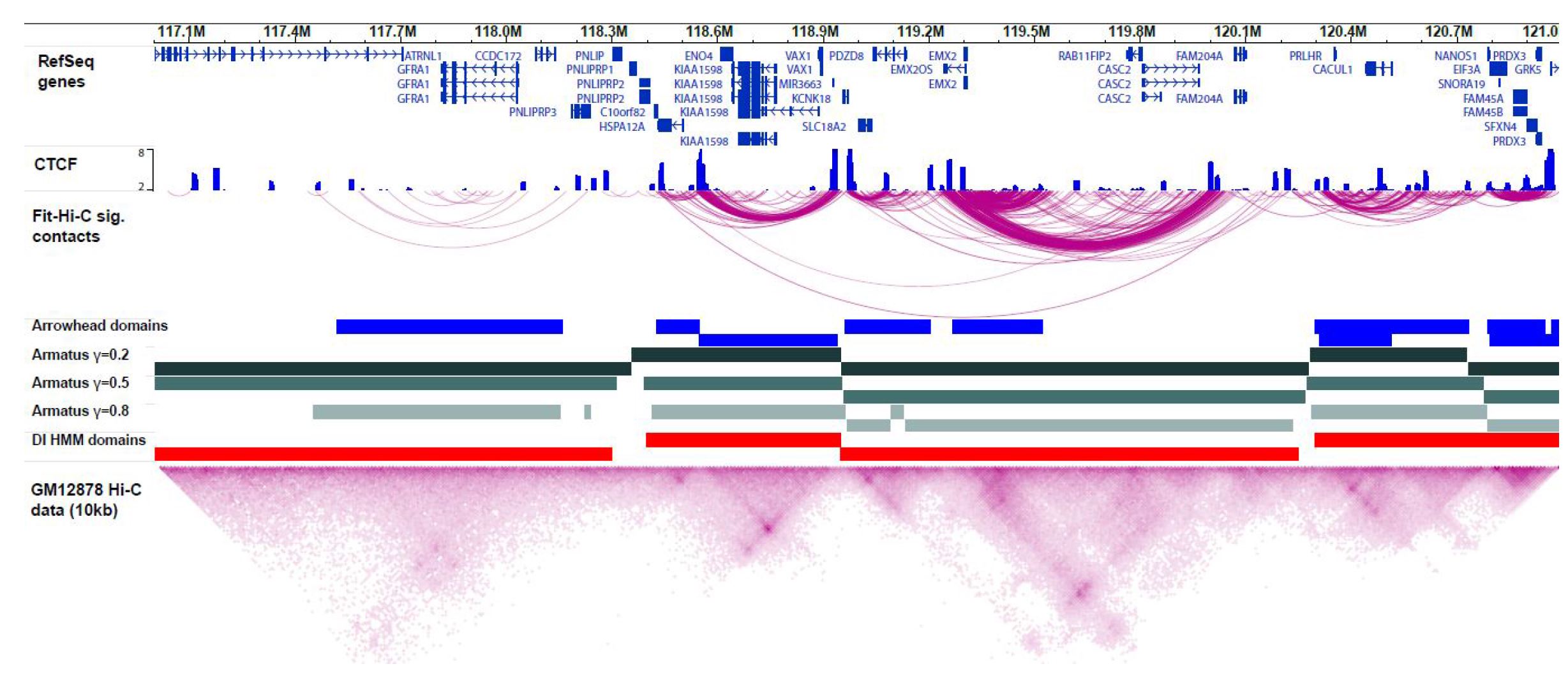
The rapidly increasing quantity of genome-wide chromosome conformation capture data presents great opportunities and challenges in the computational modeling and interpretation of the three-dimensional genome. In particular, with recent trends towards higher-resolution high-throughput chromosome conformation capture (Hi-C) data, the diversity and complexity of biological hypotheses that can be tested necessitates rigorous computational and statistical methods as well as scalable pipelines to interpret these datasets. Here we review computational tools to interpret Hi-C data, including pipelines for mapping, filtering, and normalization, and methods for confidence estimation, domain calling, visualization, and three-dimensional modeling.
Journal Website
Xinxian Deng, Wenxiu Ma, Vijay Ramani, Andrew Hill, Fan Yang, Ferhat Ay, Joel Berletch, Carl Blau, Jay Shendure, Zhijun Duan, William Noble, Christine Disteche " Bipartite structure of the inactive mouse X chromosome" Genome Biology. 16:152, 2015
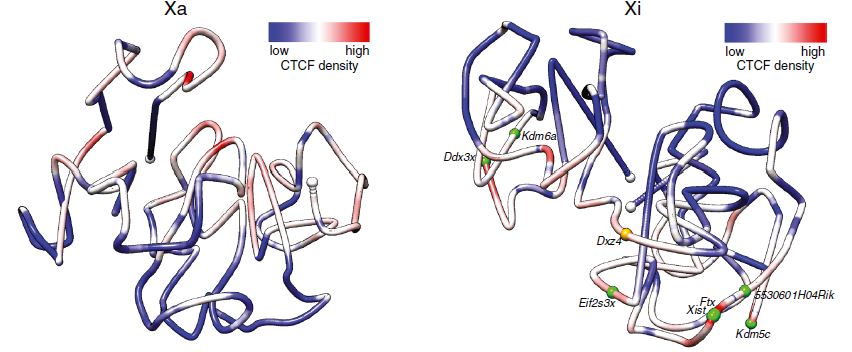
BACKGROUND: In mammals, one of the female X chromosomes and all imprinted genes are expressed exclusively from a single allele in somatic cells. To evaluate structural changes associated with allelic silencing, we have applied a recently developed Hi-C assay that uses DNase I for chromatin fragmentation to mouse F1 hybrid systems.
RESULTS: We find radically different conformations for the two female mouse X chromosomes. The inactive X has two superdomains of frequent intrachromosomal contacts separated by a boundary region. Comparison with the recently reported two-superdomain structure of the human inactive X shows that the genomic content of the superdomains differs between species, but part of the boundary region is conserved and located near the Dxz4/DXZ4 locus. In mouse, the boundary region also contains a minisatellite, Ds-TR, and both Dxz4 and Ds-TR appear to be anchored to the nucleolus. Genes that escape X inactivation do not cluster but are located near the periphery of the 3D structure, as are regions enriched in CTCF or RNA polymerase. Fewer short-range intrachromosomal contacts are detected for the inactive alleles of genes subject to X inactivation compared with the active alleles and with genes that escape X inactivation. This pattern is also evident for imprinted genes, in which more chromatin contacts are detected for the expressed allele.
CONCLUSIONS: By applying a novel Hi-C method to map allelic chromatin contacts, we discover a specific bipartite organization of the mouse inactive X chromosome that probably plays an important role in maintenance of gene silencing.
Journal Website
Rachel M. Gittelman, Enna Hun, Ferhat Ay, Jennifer Madeoy, Len Pennacchio, William S. Noble, David R. Hawkins and Joshua M. Akey "Comprehensive identification and analysis of human accelerated regulatory DNA" Genome Research, 25(9):1245-1255, 2015.
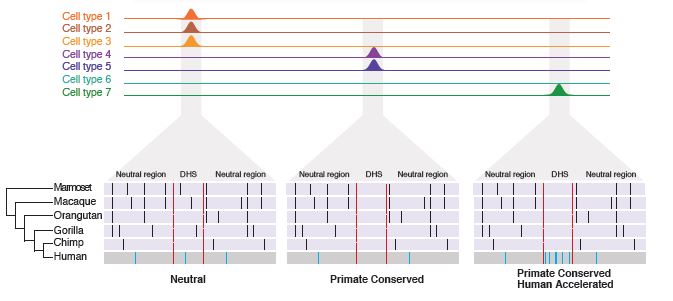
It has long been hypothesized that changes in gene regulation have played an important role in human evolution, but regulatory DNA has been much more difficult to study compared with protein-coding regions. Recent large-scale studies have created genome-scale catalogs of DNase I hypersensitive sites (DHSs), which demark potentially functional regulatory DNA. To better define regulatory DNA that has been subject to human-specific adaptive evolution, we performed comprehensive evolutionary and population genetics analyses on over 18 million DHSs discovered in 130 cell types. We identified 524 DHSs that are conserved in nonhuman primates but accelerated in the human lineage (haDHS), and estimate that 70% of substitutions in haDHSs are attributable to positive selection. Through extensive computational and experimental analyses, we demonstrate that haDHSs are often active in brain or neuronal cell types; play an important role in regulating the expression of developmentally important genes, including many transcription factors such as SOX6, POU3F2, and HOX genes; and identify striking examples of adaptive regulatory evolution that may have contributed to human-specific phenotypes. More generally, our results reveal new insights into conserved and adaptive regulatory DNA in humans and refine the set of genomic substrates that distinguish humans from their closest living primate relatives.
Journal Website
Sushmita Roy, Alireza Fotuhi Siahpirani, Deborah Chasman, Sara Knaack, Ferhat Ay, Ron Stewart, Michael Wilson, Rupa Sridharan "A predictive modeling approach for cell-line specific long-range regulatory interactions" Nucleic Acids Research, 2015.
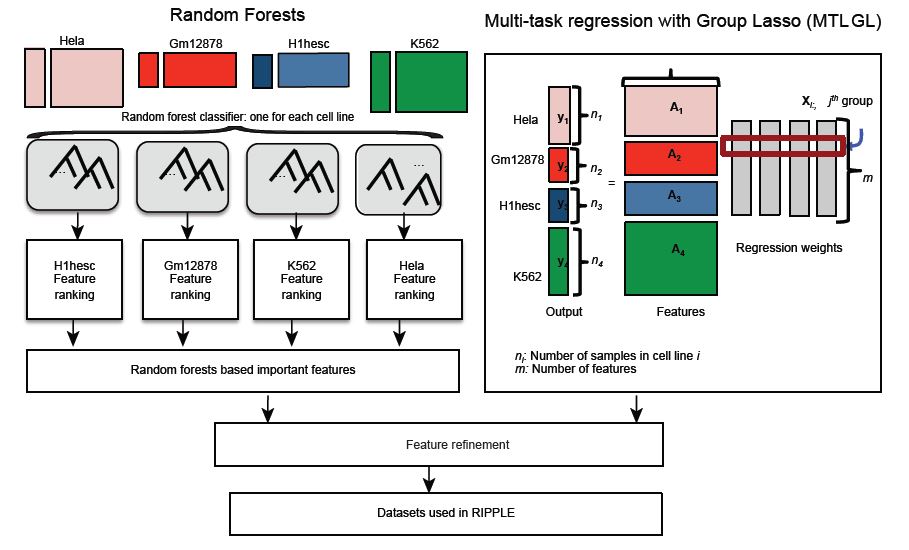
Long range regulatory interactions among distal enhancers and target genes are important for tissue-specific gene expression. Genome-scale identification of these interactions in a cell line-specific manner, especially using the fewest possible datasets, is a significant challenge. We develop a novel computational approach, Regulatory Interaction Prediction for Promoters and Long-range Enhancers (RIPPLE), that integrates published Chromosome Conformation Capture (3C) data sets with a minimal set of regulatory genomic data sets to predict enhancer-promoter interactions in a cell line-specific manner. Our results suggest that CTCF, RAD21, a general transcription factor (TBP) and activating chromatin marks are important determinants of enhancer-promoter interactions. To predict interactions in a new cell line and to generate genome-wide interaction maps, we develop an ensemble version of RIPPLE and apply it to generate interactions in five human cell lines. Computational validation of these predictions using existing ChIA-PET and Hi-C data sets showed that RIPPLE accurately predicts interactions among enhancers and promoters. Enhancer-promoter interactions tend to be organized into subnetworks representing coordinately regulated sets of genes that are enriched for specific biological processes and cis-regulatory elements. Overall, our work provides a systematic approach to predict and interpret enhancer-promoter interactions in a genome-wide cell-type specific manner using a few experimentally tractable measurements.
Journal Website
Vishnu Dileep, Ferhat Ay, Jiao Sima, Daniel L. Vera, William S. Noble, David M. Gilbert "Topologically-associating domains and their long-range contacts are established during early G1 coincident with the establishment of the replication timing program" Genome Research, 25(8):1104-1113, 2015.
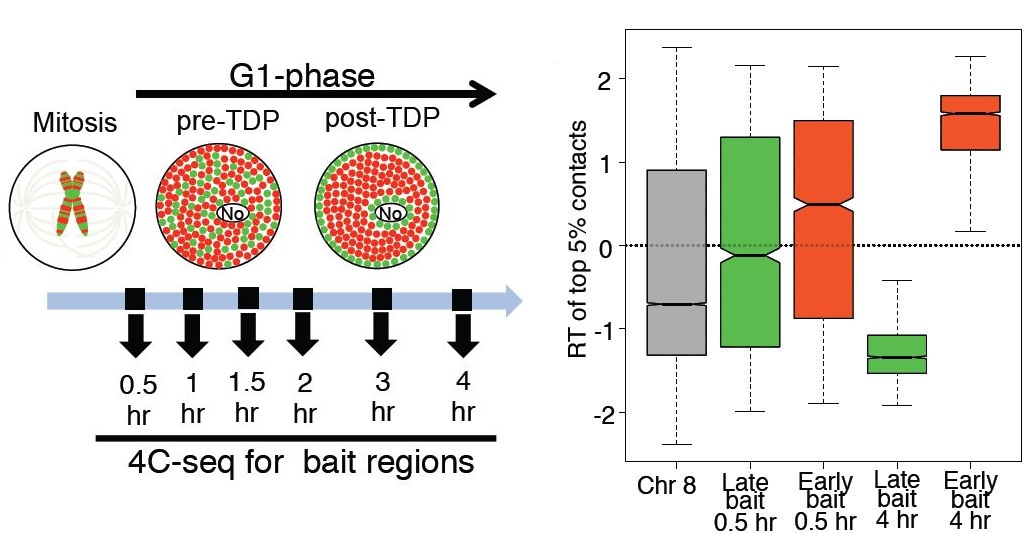
Mammalian genomes are partitioned into domains that replicate in a defined temporal order. These domains can replicate at similar times in all cell types (constitutive) or at cell type-specific times (developmental). Genome-wide chromatin conformation capture (Hi-C) has revealed sub-megabase topologically associating domains (TADs), which are the structural counterparts of replication domains. Hi-C also segregates inter-TAD contacts into defined 3D spatial compartments that align precisely to genome-wide replication timing profiles. Determinants of the replication-timing program are re-established during early G1 phase of each cell cycle and lost in G2 phase, but it is not known when TAD structure and inter-TAD contacts are re-established after their elimination during mitosis. Here, we use multiplexed 4C-seq to study dynamic changes in chromatin organization during early G1. We find that both establishment of TADs and their compartmentalization occur during early G1, within the same time frame as establishment of the replication-timing program. Once established, this 3D organization is preserved either after withdrawal into quiescence or for the remainder of interphase including G2 phase, implying 3D structure is not sufficient to maintain replication timing. Finally, we find that developmental domains are less well compartmentalized than constitutive domains and display chromatin properties that distinguish them from early and late constitutive domains. Overall, this study uncovers a strong connection between chromatin re-organization during G1, establishment of replication timing, and its developmental control.
Journal Website
[Nucleic Acids Research] Accurate identification of centromere locations in yeast genomes using Hi-C
Nelle Varoquaux, Ivan Liachko, Ferhat Ay, Joshua N. Burton, Jay Shendure, Maitreya J. Dunham, Jean-Philippe Vert, William S. Noble "Accurate identification of centromere locations in yeast genomes using Hi-C" Nucleic Acids Research, 3(11):5331-5339, 2015.
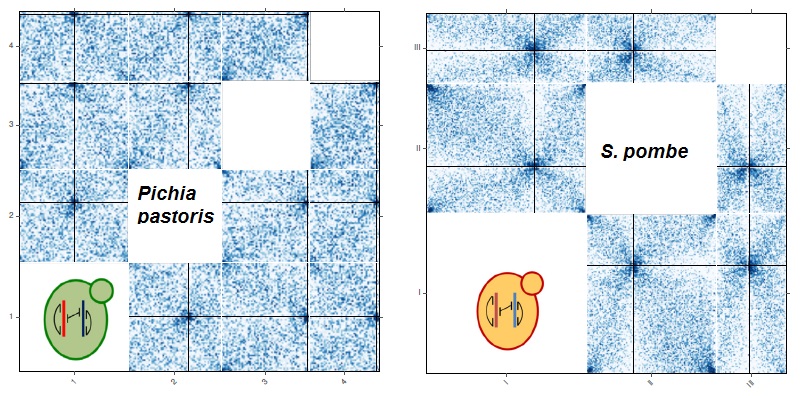
Centromeres are essential for proper chromosome segregation. Despite extensive research, centromere locations in yeast genomes remain difficult to infer, and in most species they are still unknown. Recently, the chromatin conformation capture assay, Hi-C, has been re-purposed for diverse applications, including de novo genome assembly, deconvolution of metagenomic samples and inference of centromere locations. We describe a method, Centurion, that jointly infers the locations of all centromeres in a single genome from Hi-C data by exploiting the centromeres' tendency to cluster in three-dimensional space. We first demonstrate the accuracy of Centurion in identifying known centromere locations from high coverage Hi-C data of budding yeast and a human malaria parasite. We then use Centurion to infer centromere locations in 14 yeast species. Across all microbes that we consider, Centurion predicts 89% of centromeres within 5 kb of their known locations. We also demonstrate the robustness of the approach in datasets with low sequencing depth. Finally, we predict centromere coordinates for six yeast species that currently lack centromere annotations. These results show that Centurion can be used for centromere identification for diverse species of yeast and possibly other microorganisms.
Journal Website
Maxwell W. Libbrecht, Ferhat Ay, Michael M. Hoffman, David M. Gilbert, Jeffrey A. Bilmes, William S. Noble "Joint annotation of chromatin state and chromatin conformation reveals relationships among domain types and identifies domains of cell type-specific expression" Genome Research, 25(4):544-557, 2015.
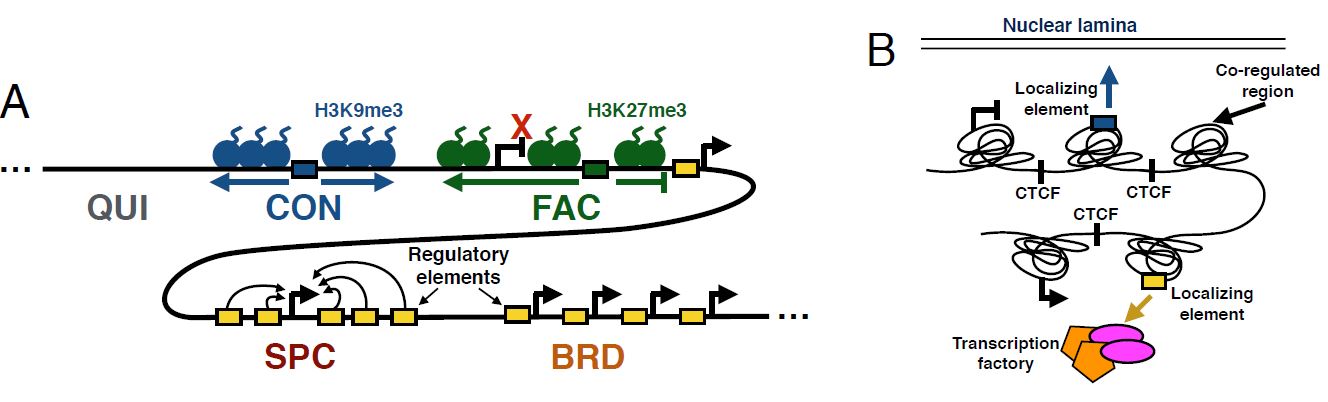
The genomic neighborhood of a gene influences its activity, a behavior that is attributable in part to domain-scale regulation. Previous genomic studies have identified many types of regulatory domains. However, due to the difficulty of integrating genomics data sets, the relationships among these domain types are poorly understood. Semi-automated genome annotation (SAGA) algorithms facilitate human interpretation of heterogeneous collections of genomics data by simultaneously partitioning the human genome and assigning labels to the resulting genomic segments. However, existing SAGA methods cannot integrate inherently pairwise chromatin conformation data. We developed a new computational method, called graph-based regularization (GBR), for expressing a pairwise prior that encourages certain pairs of genomic loci to receive the same label in a genome annotation. We used GBR to exploit chromatin conformation information during genome annotation by encouraging positions that are close in 3D to occupy the same type of domain. Using this approach, we produced a model of chromatin domains in eight human cell types, thereby revealing the relationships among known domain types. Through this model, we identified clusters of tightly regulated genes expressed in only a small number of cell types, which we term "specific expression domains." We found that domain boundaries marked by promoters and CTCF motifs are consistent between cell types even when domain activity changes. Finally, we showed that GBR can be used to transfer information from well-studied cell types to less well-characterized cell types during genome annotation, making it possible to produce high-quality annotations of the hundreds of cell types with limited available data.
Journal Website
Ferhat Ay, Thanh H. Vu, Michael J. Zeitz, Nelle Varoquaux, Jan E. Carette, Jean-Philippe Vert, Andrew R. Hoffman, William S. Noble "Identifying multi-locus chromatin contacts in human cells using tethered multiple 3C" BMC Genomics, 16:121, 2015.
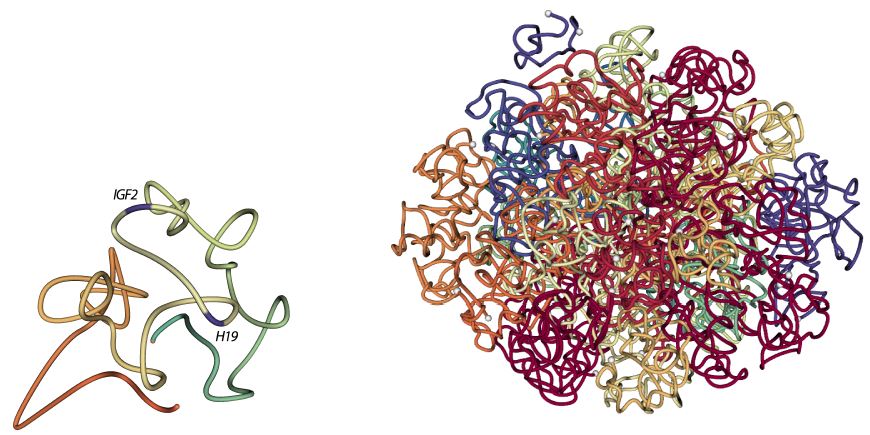
BACKGROUND: Several recently developed experimental methods, each an extension of the chromatin conformation capture (3C) assay, have enabled the genome-wide profiling of chromatin contacts between pairs of genomic loci in 3D. Especially in complex eukaryotes, data generated by these methods, coupled with other genome-wide datasets, demonstrated that non-random chromatin folding correlates strongly with cellular processes such as gene expression and DNA replication.
RESULTS: We describe a genome architecture assay, tethered multiple 3C (TM3C), that maps genome-wide chromatin contacts via a simple protocol of restriction enzyme digestion and religation of fragments upon agarose gel beads followed by paired-end sequencing. In addition to identifying contacts between pairs of loci, TM3C enables identification of contacts among more than two loci simultaneously. We use TM3C to assay the genome architectures of two human cell lines: KBM7, a near-haploid chronic leukemia cell line, and NHEK, a normal diploid human epidermal keratinocyte cell line. We confirm that the contact frequency maps produced by TM3C exhibit features characteristic of existing genome architecture datasets, including the expected scaling of contact probabilities with genomic distance, megabase scale chromosomal compartments and sub-megabase scale topological domains. We also confirm that TM3C captures several known cell type-specific contacts, ploidy shifts and translocations, such as Philadelphia chromosome formation (Ph+) in KBM7. We confirm a subset of the triple contacts involving the IGF2-H19 imprinting control region (ICR) using PCR analysis for KBM7 cells. Our genome-wide analysis of pairwise and triple contacts demonstrates their preference for linking open chromatin regions to each other and for linking regions with higher numbers of DNase hypersensitive sites (DHSs) to each other. For near-haploid KBM7 cells, we infer whole genome 3D models that exhibit clustering of small chromosomes with each other and large chromosomes with each other, consistent with previous studies of the genome architectures of other human cell lines.
CONCLUSION: TM3C is a simple protocol for ascertaining genome architecture and can be used to identify simultaneous contacts among three or four loci. Application of TM3C to a near-haploid human cell line revealed large-scale features of chromosomal organization and multi-way chromatin contacts that preferentially link regions of open chromatin.
Journal Website
Wenxiu Ma, Ferhat Ay, Choli Lee, Gunhan Gulsoy, Xinxian Deng, Savannah Cook, Jennifer Hesson, Christopher Cavanaugh, Carol B. Ware, Anton Krumm, Jay Shendure, C. Anthony Blau, Christine M. Disteche, William S. Noble, ZhiJun Duan "Fine-scale chromatin interaction maps reveal the cis-regulatory landscape of human lincRNA genes" Nature Methods, 12(1):71-78, 2015.
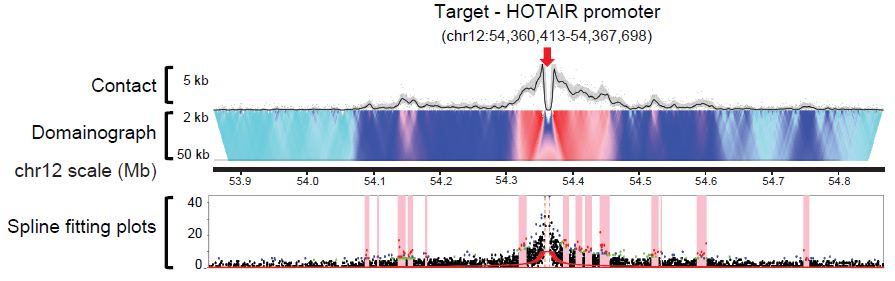
High-throughput methods based on chromosome conformation capture have greatly advanced our understanding of the three-dimensional (3D) organization of genomes but are limited in resolution by their reliance on restriction enzymes. Here we describe a method called DNase Hi-C for comprehensively mapping global chromatin contacts. DNase Hi-C uses DNase I for chromatin fragmentation, leading to greatly improved efficiency and resolution over that of Hi-C. Coupling this method with DNA-capture technology provides a high-throughput approach for targeted mapping of fine-scale chromatin architecture. We applied targeted DNase Hi-C to characterize the 3D organization of 998 large intergenic noncoding RNA (lincRNA) promoters in two human cell lines. Our results revealed that expression of lincRNAs is tightly controlled by complex mechanisms involving both super-enhancers and the Polycomb repressive complex. Our results provide the first glimpse of the cell type-specific 3D organization of lincRNA genes.
Journal Website
Ferhat Ay, Evelien M. Bunnik, Nelle Varoquaux, Jean-Philippe Vert, William Stafford Noble, Karine G. Le Roch "Multiple dimensions of epigenetic gene regulation in the malaria parasite Plasmodium falciparum" Bioessays, 37(2):182-194, 2015.
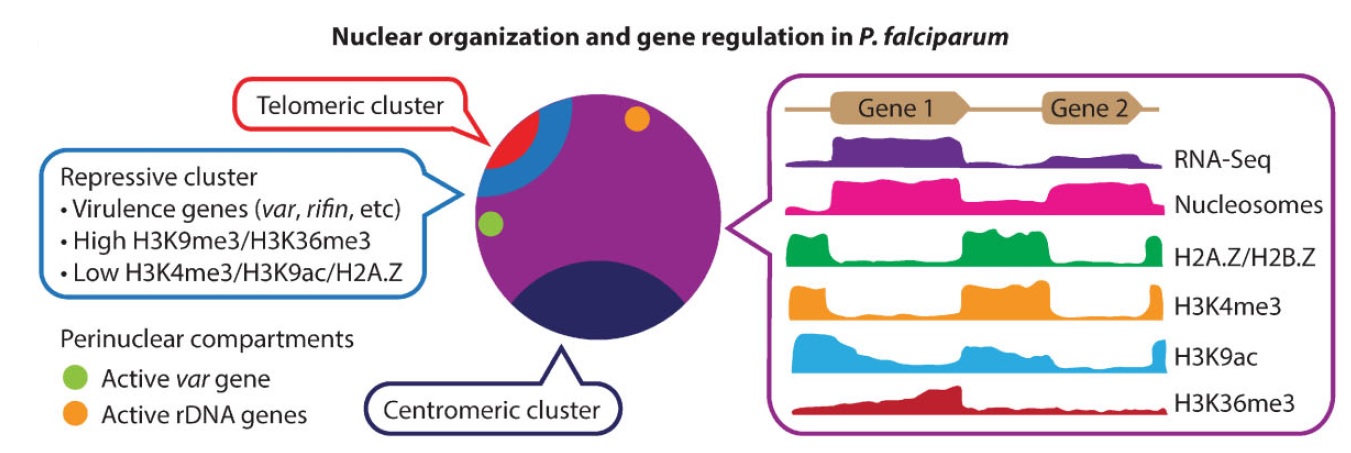
Plasmodium falciparum is the most deadly human malarial parasite, responsible for an estimated 207 million cases of disease and 627,000 deaths in 2012. Recent studies reveal that the parasite actively regulates a large fraction of its genes throughout its replicative cycle inside human red blood cells and that epigenetics plays an important role in this precise gene regulation. Here, we discuss recent advances in our understanding of three aspects of epigenetic regulation in P. falciparum: changes in histone modifications, nucleosome occupancy and the three-dimensional genome structure. We compare these three aspects of the P. falciparum epigenome to those of other eukaryotes, and show that large-scale compartmentalization is particularly important in determining histone decomposition and gene regulation in P. falciparum. We conclude by presenting a gene regulation model for P. falciparum that combines the described epigenetic factors, and by discussing the implications of this model for the future of malaria research.
Journal Wesbite
2014
Nelle Varoquaux, Ferhat Ay, Jean-Philippe Vert, William Stafford Noble "A statistical approach for inferring the 3D structure of the genome" Bioinformatics 30(12): i26-i33, 2014.

MOTIVATION: Recent technological advances allow the measurement, in a single Hi-C experiment, of the frequencies of physical contacts among pairs of genomic loci at a genome-wide scale. The next challenge is to infer, from the resulting DNA-DNA contact maps, accurate 3D models of how chromosomes fold and fit into the nucleus. Many existing inference methods rely on multidimensional scaling (MDS), in which the pairwise distances of the inferred model are optimized to resemble pairwise distances derived directly from the contact counts. These approaches, however, often optimize a heuristic objective function and require strong assumptions about the biophysics of DNA to transform interaction frequencies to spatial distance, and thereby may lead to incorrect structure reconstruction.
METHODS: We propose a novel approach to infer a consensus 3D structure of a genome from Hi-C data. The method incorporates a statistical model of the contact counts, assuming that the counts between two loci follow a Poisson distribution whose intensity decreases with the physical distances between the loci. The method can automatically adjust the transfer function relating the spatial distance to the Poisson intensity and infer a genome structure that best explains the observed data.
RESULTS: We compare two variants of our Poisson method, with or without optimization of the transfer function, to four different MDS-based algorithms-two metric MDS methods using different stress functions, a non-metric version of MDS and ChromSDE, a recently described, advanced MDS method-on a wide range of simulated datasets. We demonstrate that the Poisson models reconstruct better structures than all MDS-based methods, particularly at low coverage and high resolution, and we highlight the importance of optimizing the transfer function. On publicly available Hi-C data from mouse embryonic stem cells, we show that the Poisson methods lead to more reproducible structures than MDS-based methods when we use data generated using different restriction enzymes, and when we reconstruct structures at different resolutions.
AVAILABILITY AND IMPLEMENTATION: A Python implementation of the proposed method is available at http://cbio.ensmp.fr/pastis.
Journal Website
Ferhat Ay, Evelien M. Bunnik, Nelle Varoquaux, Sebastiaan M. Bol, Jacques Prudhomme, Jean-Philippe Vert, William Stafford Noble, Karine G. Le Roch "Three-dimensional modeling of the P. falciparum genome during the erythrocytic cycle reveals a strong connection between genome architecture and gene expression" Genome Research, 24: 974-988, 2014.
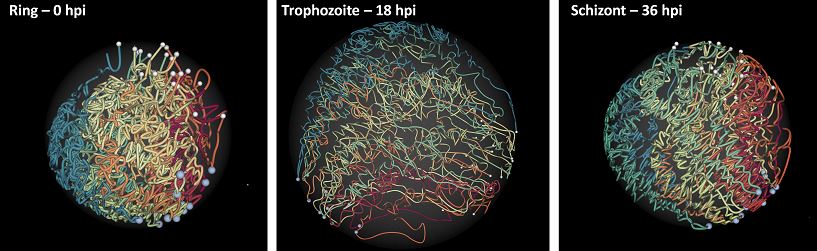
The development of the human malaria parasite Plasmodium falciparum is controlled by coordinated changes in gene expression throughout its complex life cycle, but the corresponding regulatory mechanisms are incompletely understood. To study the relationship between genome architecture and gene regulation in Plasmodium, we assayed the genome architecture of P. falciparum at three time points during its erythrocytic (asexual) cycle. Using chromosome conformation capture coupled with next-generation sequencing technology (Hi-C), we obtained high-resolution chromosomal contact maps, which we then used to construct a consensus three-dimensional genome structure for each time point. We observed strong clustering of centromeres, telomeres, ribosomal DNA, and virulence genes, resulting in a complex architecture that cannot be explained by a simple volume exclusion model. Internal virulence gene clusters exhibit domain-like structures in contact maps, suggesting that they play an important role in the genome architecture. Midway during the erythrocytic cycle, at the highly transcriptionally active trophozoite stage, the genome adopts a more open chromatin structure with increased chromosomal intermingling. In addition, we observed reduced expression of genes located in spatial proximity to the repressive subtelomeric center, and colocalization of distinct groups of parasite-specific genes with coordinated expression profiles. Overall, our results are indicative of a strong association between the P. falciparum spatial genome organization and gene expression. Understanding the molecular processes involved in genome conformation dynamics could contribute to the discovery of novel antimalarial strategies.
Journal Website
Ferhat Ay, Timothy L. Bailey, William S. Noble "Fit-Hi-C: Statistical confidence estimation for Hi-C data reveals regulatory chromatin contacts" Genome Research, 24: 999-1011, 2014.
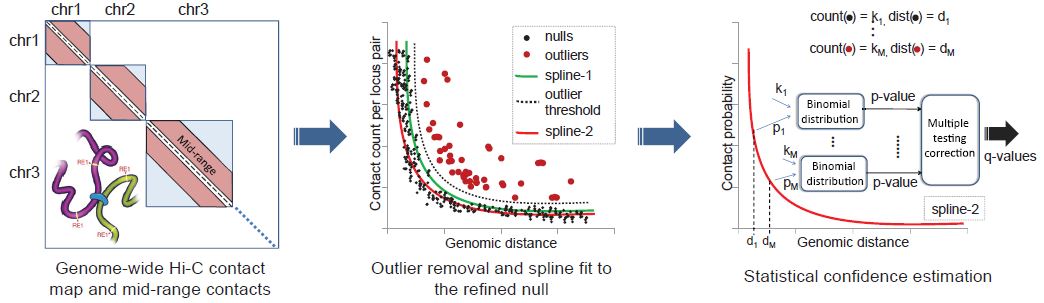
Our current understanding of how DNA is packed in the nucleus is most accurate at the fine scale of individual nucleosomes and at the large scale of chromosome territories. However, accurate modeling of DNA architecture at the intermediate scale of ∼50 kb-10 Mb is crucial for identifying functional interactions among regulatory elements and their target promoters. We describe a method, Fit-Hi-C, that assigns statistical confidence estimates to mid-range intra-chromosomal contacts by jointly modeling the random polymer looping effect and previously observed technical biases in Hi-C data sets. We demonstrate that our proposed approach computes accurate empirical null models of contact probability without any distribution assumption, corrects for binning artifacts, and provides improved statistical power relative to a previously described method. High-confidence contacts identified by Fit-Hi-C preferentially link expressed gene promoters to active enhancers identified by chromatin signatures in human embryonic stem cells (ESCs), capture 77% of RNA polymerase II-mediated enhancer-promoter interactions identified using ChIA-PET in mouse ESCs, and confirm previously validated, cell line-specific interactions in mouse cortex cells. We observe that insulators and heterochromatin regions are hubs for high-confidence contacts, while promoters and strong enhancers are involved in fewer contacts. We also observe that binding peaks of master pluripotency factors such as NANOG and POU5F1 are highly enriched in high-confidence contacts for human ESCs. Furthermore, we show that pairs of loci linked by high-confidence contacts exhibit similar replication timing in human and mouse ESCs and preferentially lie within the boundaries of topological domains for human and mouse cell lines.
Journal Website
2013
Michael J. Zeitz, Paula L. Lerner, Ferhat Ay, Eric V. Nostrand, Julia D. Heidmann, William S. Noble, Andrew R. Hoffman "Implications of COMT long-range interactions on the phenotypic variability of 22q11.2 deletion syndrome" Nucleus, 4(6): 487-493, 2013.
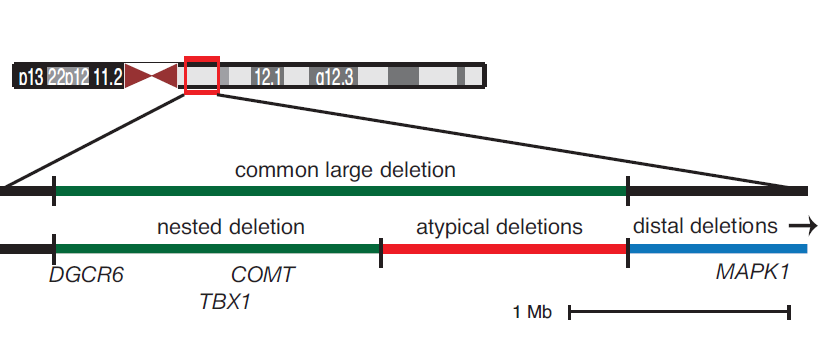
22q11.2 deletion syndrome (22q11DS) results from a hemizygous microdeletion on chromosome 22 and is characterized by extensive phenotypic variability. Penetrance of signs, including congenital heart, craniofacial, and neurobehavioral abnormalities, varies widely and is not well correlated with genotype. The three-dimensional structure of the genome may help explain some of this variability. The physical interaction profile of a given gene locus with other genetic elements, such as enhancers and co-regulated genes, contributes to its regulation. Thus, it is possible that regulatory interactions with elements outside the deletion region are disrupted in the disease state and modulate the resulting spectrum of symptoms. COMT, a gene within the commonly deleted ~3 Mb region has been implicated as a contributor to the neurological features frequently found in 22q11DS patients. We used this locus as bait in a 4C-seq experiment to investigate genome-wide interaction profiles in B lymphocyte and fibroblast cell lines derived from both 22q11DS and unaffected individuals. All normal B lymphocyte lines displayed local, conserved chromatin looping interactions with regions that are lost in atypical and distal deletions, which may mediate similarities between typical, atypical, and distal 22q11 deletion phenotypes. There are also distinct clusterings of cis interactions based on disease state. We identified regions of differential trans interactions present in normal, and lost in deletion-carrying, B lymphocyte cell lines. This data suggests that hemizygous chromosomal deletions such as 22q11DS can have widespread effects on chromatin organization, and may contribute to the inherent phenotypic variability.
Journal Website
Michael J. Zeitz, Ferhat Ay, Julia D. Heidmann, Paula L. Lerner, William S. Noble, Brandon N. Steelman, Andrew R. Hoffman "Genomic Interaction Profiles in Breast Cancer Reveal Altered Chromatin Architecture" PLoS ONE, 8(9):e73974, 2013.
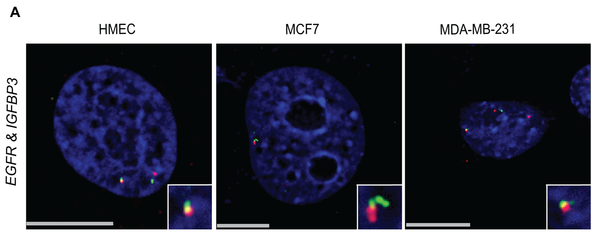
Gene transcription can be regulated by remote enhancer regions through chromosome looping either in cis or in trans. Cancer cells are characterized by wholesale changes in long-range gene interactions, but the role that these long-range interactions play in cancer progression and metastasis is not well understood. In this study, we used IGFBP3, a gene involved in breast cancer pathogenesis, as bait in a 4C-seq experiment comparing normal breast cells (HMEC) with two breast cancer cell lines (MCF7, an ER positive cell line, and MDA-MB-231, a triple negative cell line). The IGFBP3 long-range interaction profile was substantially altered in breast cancer. Many interactions seen in normal breast cells are lost and novel interactions appear in cancer lines. We found that in HMEC, the breast carcinoma amplified sequence gene family (BCAS) 1-4 were among the top 10 most significantly enriched regions of interaction with IGFBP3. 3D-FISH analysis indicated that the translocation-prone BCAS genes, which are located on chromosomes 1, 17, and 20, are in close physical proximity with IGFBP3 and each other in normal breast cells. We also found that epidermal growth factor receptor (EGFR), a gene implicated in tumorigenesis, interacts significantly with IGFBP3 and that this interaction may play a role in their regulation. Breakpoint analysis suggests that when an IGFBP3 interacting region undergoes a translocation an additional interaction detectable by 4C is gained. Overall, our data from multiple lines of evidence suggest an important role for long-range chromosomal interactions in the pathogenesis of cancer.
Journal Website
2012
David Marbach, Sushmita. Roy, Ferhat Ay, Patrick E. Meyer, Rogerio Candeias, Tamer Kahveci, Christopher A. Bristow and Manolis Kellis "Predictive regulatory models in Drosophila Melanogaster by integrative inference of transcriptional networks" Genome Research, gr.127191.111, 2012.
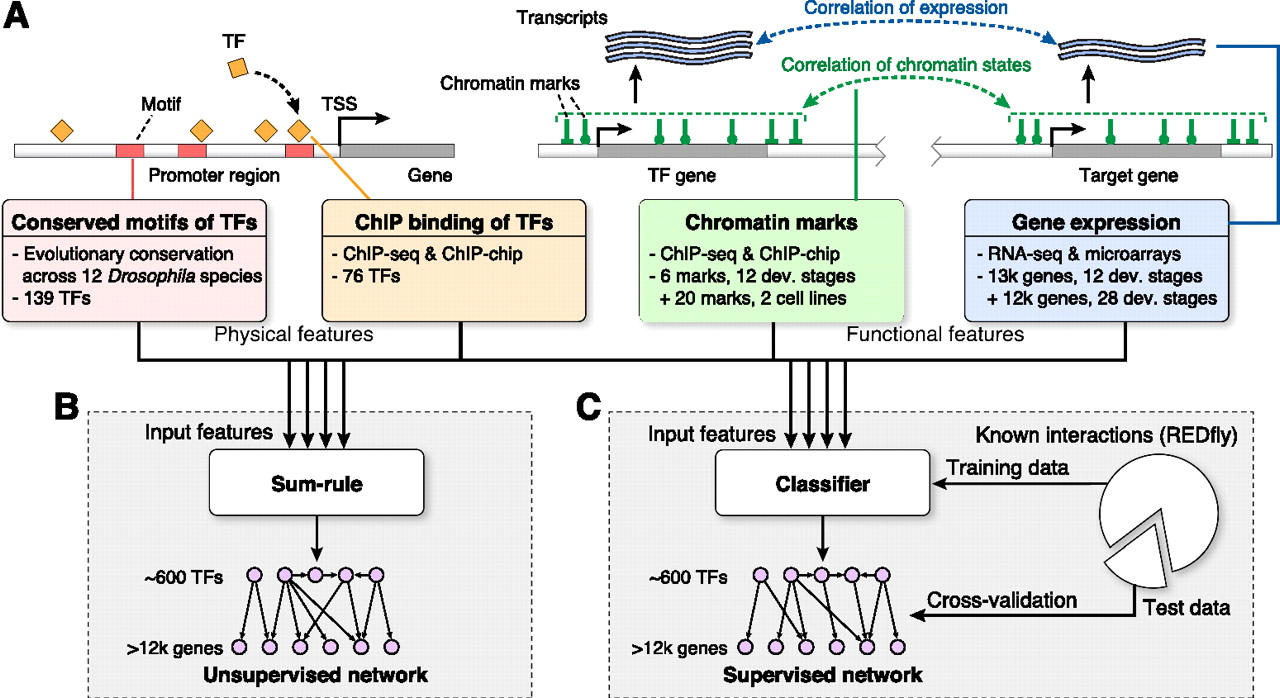
Gaining insights on gene regulation from large-scale functional data sets is a grand challenge in systems biology. In this article, we develop and apply methods for transcriptional regulatory network inference from diverse functional genomics data sets and demonstrate their value for gene function and gene expression prediction. We formulate the network inference problem in a machine-learning framework and use both supervised and unsupervised methods to predict regulatory edges by integrating transcription factor (TF) binding, evolutionarily conserved sequence motifs, gene expression, and chromatin modification data sets as input features. Applying these methods to Drosophila melanogaster, we predict ∼300,000 regulatory edges in a network of ∼600 TFs and 12,000 target genes. We validate our predictions using known regulatory interactions, gene functional annotations, tissue-specific expression, protein-protein interactions, and three-dimensional maps of chromosome conformation. We use the inferred network to identify putative functions for hundreds of previously uncharacterized genes, including many in nervous system development, which are independently confirmed based on their tissue-specific expression patterns. Last, we use the regulatory network to predict target gene expression levels as a function of TF expression, and find significantly higher predictive power for integrative networks than for motif or ChIP-based networks. Our work reveals the complementarity between physical evidence of regulatory interactions (TF binding, motif conservation) and functional evidence (coordinated expression or chromatin patterns) and demonstrates the power of data integration for network inference and studies of gene regulation at the systems level.
Journal Wesbite
Ferhat Ay, Michael Dang, Tamer Kahveci "Metabolic Network Alignment in Large Scale by Network Compression" BMC Bioinformatics, 13(Suppl 3):S2, 2012.
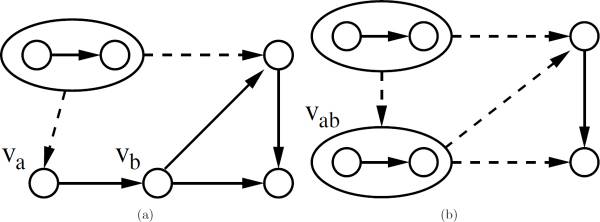
Metabolic network alignment is a system scale comparative analysis that discovers important similarities and differences across different metabolisms and organisms. Although the problem of aligning metabolic networks has been considered in the past, the computational complexity of the existing solutions has so far limited their use to moderately sized networks. In this paper, we address the problem of aligning two metabolic networks, particularly when both of them are too large to be dealt with using existing methods. We develop a generic framework that can significantly improve the scale of the networks that can be aligned in practical time. Our framework has three major phases, namely the compression phase, the alignment phase and the refinement phase. For the first phase, we develop an algorithm which transforms the given networks to a compressed domain where they are summarized using fewer nodes, termed supernodes, and interactions. In the second phase, we carry out the alignment in the compressed domain using an existing network alignment method as our base algorithm. This alignment results in supernode mappings in the compressed domain, each of which are smaller instances of network alignment problem. In the third phase, we solve each of the instances using the base alignment algorithm to refine the alignment results. We provide a user defined parameter to control the number of compression levels which generally determines the tradeoff between the quality of the alignment versus how fast the algorithm runs. Our experiments on the networks from KEGG pathway database demonstrate that the compression method we propose reduces the sizes of metabolic networks by almost half at each compression level which provides an expected speedup of more than an order of magnitude. We also observe that the alignments obtained by only one level of compression capture the original alignment results with high accuracy. Together, these suggest that our framework results in alignments that are comparable to existing algorithms and can do this with practical resource utilization for large scale networks that existing algorithms could not handle. As an example of our method's performance in practice, the alignment of organism-wide metabolic networks of human (1615 reactions) and mouse (1600 reactions) was performed under three minutes by only using a single level of compression.
Journal Website
2011
Ferhat Ay, Manolis Kellis, Tamer Kahveci "SubMAP: Aligning metabolic pathways with subnetwork mappings" Journal of Computational Biology (JCB), 18(3):1-17, 2011.
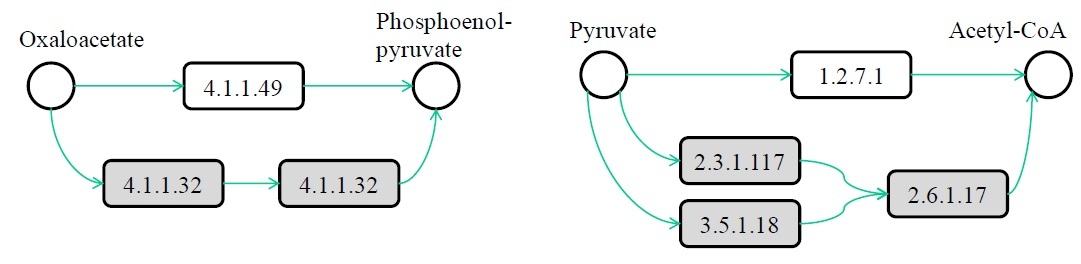
We consider the problem of aligning two metabolic pathways. Unlike traditional approaches, we do not restrict the alignment to one-to-one mappings between the molecules (nodes) of the input pathways (graphs). We follow the observation that, in nature, different organisms can perform the same or similar functions through different sets of reactions and molecules. The number and the topology of the molecules in these alternative sets often vary from one organism to another. With the motivation that an accurate biological alignment should be able to reveal these functionally similar molecule sets across different species, we develop an algorithm that first measures the similarities between different nodes using a mixture of homology and topological similarity. We combine the two metrics by employing an eigenvalue formulation. We then search for an alignment between the two input pathways that maximizes a similarity score, evaluated as the sum of the similarities of the mapped subnetworks of size at most a given integer k, and also does not contain any conflicting mappings. Here we prove that this maximization is NP-hard by a reduction from the maximum weight independent set (MWIS) problem. We then convert our problem to an instance of MWIS and use an efficient vertex-selection strategy to extract the mappings that constitute our alignment. We name our algorithm SubMAP (Subnetwork Mappings in Alignment of Pathways). We evaluate its accuracy and performance on real datasets. Our empirical results demonstrate that SubMAP can identify biologically relevant mappings that are missed by traditional alignment methods. Furthermore, we observe that SubMAP is scalable for metabolic pathways of arbitrary topology, including searching for a query pathway of size 70 against the complete KEGG database of 1,842 pathways. Implementation in C++ is available at http://bioinformatics.cise.ufl.edu/SubMAP.html.
Journal Website
2010
The modENCODE Consortium, Sushmita Roy*, Jason Ernst*, Peter V. Kharchenko*, Pouya Kheradpour*, Nicolas Negre*, Matthew L. Eaton*, Jane M. Landolin*, Christopher A. Bristow*, Lijia Ma*, Michael F. Lin*, Stefan Washietl*, Bradley I. Arshinoff*, Ferhat Ay*, Patrick E. Meyer*, Nicolas Robine*, Nicole L. Washington*, Luisa Di Stefano*, et. al., Susan E. Celniker, Steven Henikoff, Gary H. Karpen, Eric C. Lai, David M. MacAlpine, Lincoln D. Stein, Kevin P. White and Manolis Kellis * =contributors, co-first authors "Identification of Functional Elements and Regulatory Circuits by Drosophila modENCODE" Science, 330(6012):1787-1797, 2010.
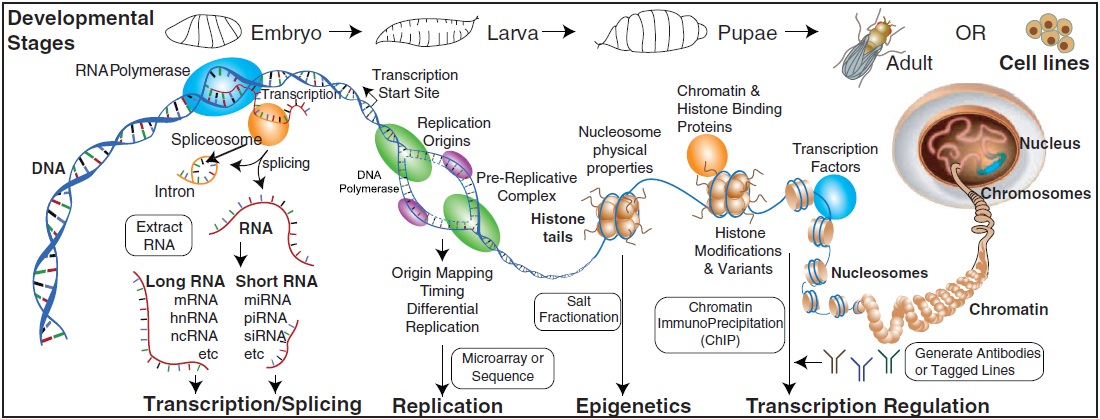
To gain insight into how genomic information is translated into cellular and developmental programs, the Drosophila model organism Encyclopedia of DNA Elements (modENCODE) project is comprehensively mapping transcripts, histone modifications, chromosomal proteins, transcription factors, replication proteins and intermediates, and nucleosome properties across a developmental time course and in multiple cell lines. We have generated more than 700 data sets and discovered protein-coding, noncoding, RNA regulatory, replication, and chromatin elements, more than tripling the annotated portion of the Drosophila genome. Correlated activity patterns of these elements reveal a functional regulatory network, which predicts putative new functions for genes, reveals stage- and tissue-specific regulators, and enables gene-expression prediction. Our results provide a foundation for directed experimental and computational studies in Drosophila and related species and also a model for systematic data integration toward comprehensive genomic and functional annotation.
Journal Website
2009
Ferhat Ay, Fei Xu, Tamer Kahveci "Scalable steady state analysis of boolean biological regulatory networks" PLoS ONE 4(12): e7992, 2009.
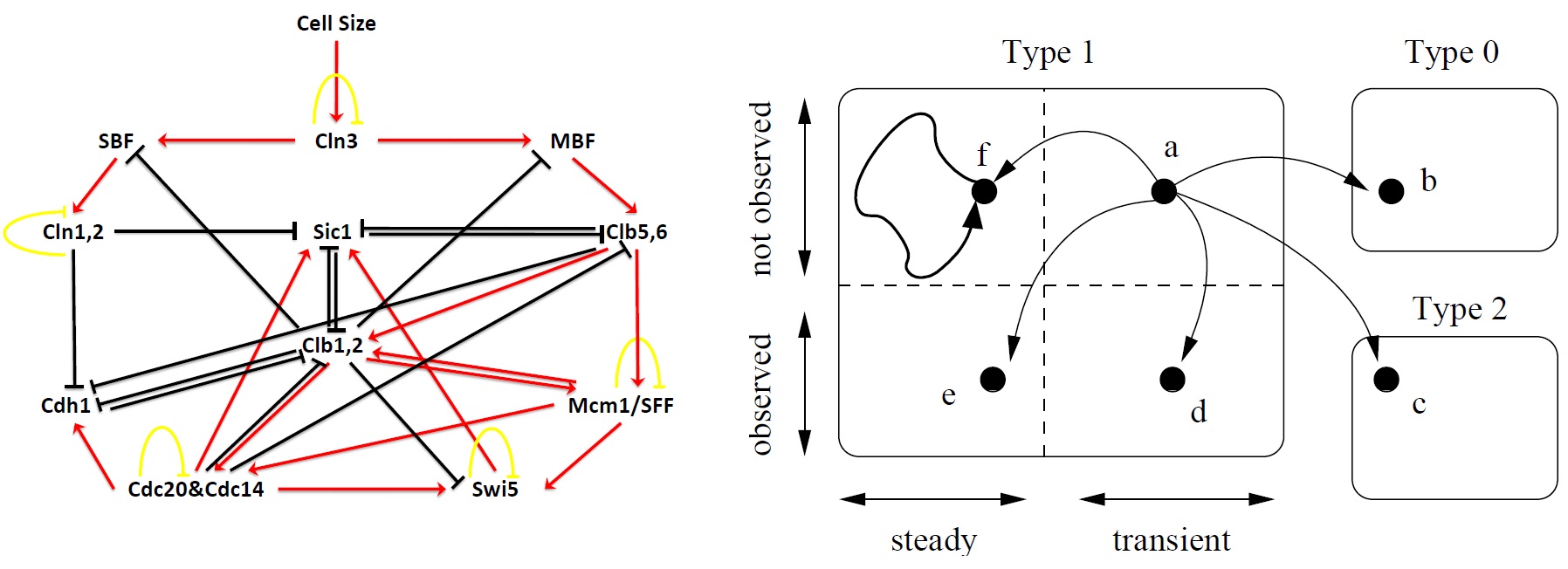
BACKGROUND: Computing the long term behavior of regulatory and signaling networks is critical in understanding how biological functions take place in organisms. Steady states of these networks determine the activity levels of individual entities in the long run. Identifying all the steady states of these networks is difficult due to the state space explosion problem.
METHODOLOGY: In this paper, we propose a method for identifying all the steady states of Boolean regulatory and signaling networks accurately and efficiently. We build a mathematical model that allows pruning a large portion of the state space quickly without causing any false dismissals. For the remaining state space, which is typically very small compared to the whole state space, we develop a randomized traversal method that extracts the steady states. We estimate the number of steady states, and the expected behavior of individual genes and gene pairs in steady states in an online fashion. Also, we formulate a stopping criterion that terminates the traversal as soon as user supplied percentage of the results are returned with high confidence.
CONCLUSIONS: This method identifies the observed steady states of boolean biological networks computationally. Our algorithm successfully reported the G1 phases of both budding and fission yeast cell cycles. Besides, the experiments suggest that this method is useful in identifying co-expressed genes as well. By analyzing the steady state profile of Hedgehog network, we were able to find the highly co-expressed gene pair GL1-SMO together with other such pairs.
Journal Website
[Computational Biology] A fast and accurate algorithm for comparative analysis of metabolic pathways
Ferhat Ay, Tamer Kahveci, Valerie de Crecy-Lagard "A fast and accurate algorithm for comparative analysis of metabolic pathways" Computational Biology (JBCB) 2009 Jun;7(3):389-428.
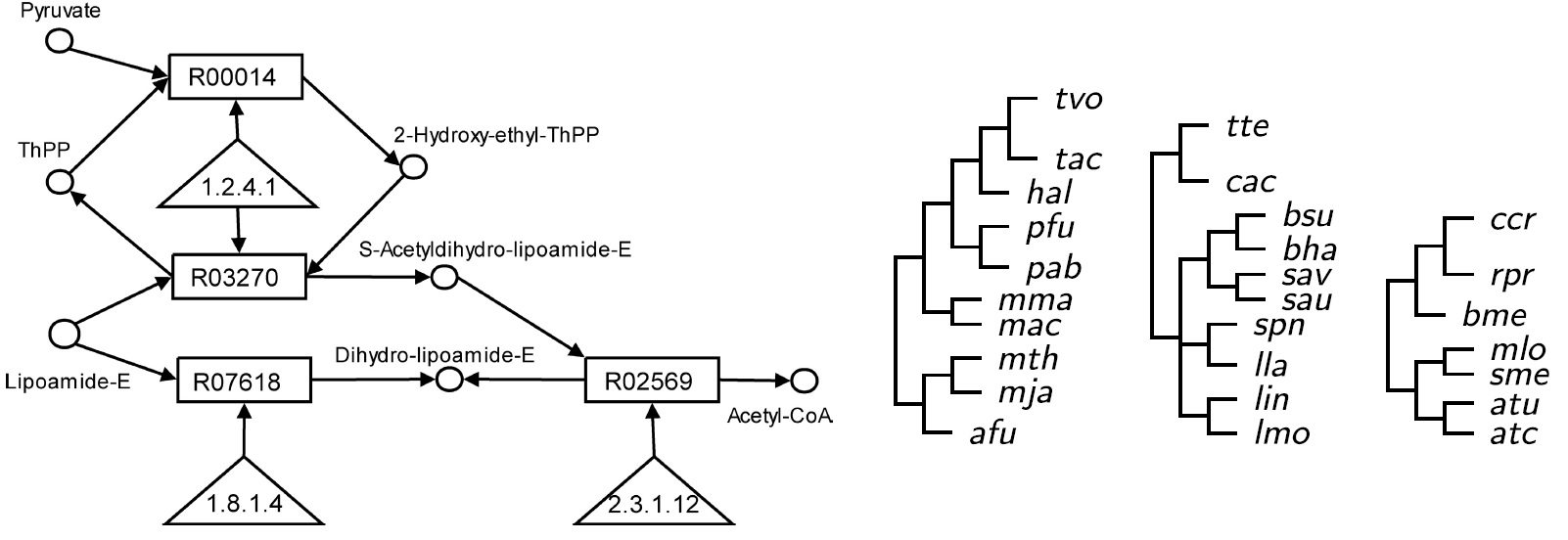
Pathways show how different biochemical entities interact with one another to perform vital functions for the survival of an organism. Comparative analysis of pathways is crucial in identifying functional similarities that are difficult to identify by comparing individual entities that build up these pathways. When interacting entities are of single type, the problem of identifying similarities by aligning the pathways can be reduced to graph isomorphism problem. For pathways with varying types of entities such as metabolic pathways, alignment problem is even more challenging. In order to simplify this problem, existing methods often reduce metabolic pathways to graphs with restricted topologies and single type of nodes. However, these abstractions reduce the relevance of the alignment significantly as they cause losses in the information content. In this paper, we describe an algorithm to solve the pairwise alignment problem for metabolic pathways. A distinguishing feature of our method is that it aligns different types of entities, such as enzymes, reactions and compounds. Also, our approach is free of any abstraction in modeling the pathways. We pursue the intuition that both pairwise similarities of entities (homology) and the organization of their interactions (topology) are important for metabolic pathway alignment. In our algorithm, we account for both by creating an eigenvalue problem for each entity type. We enforce the consistency while combining the alignments of different entity types by considering the reachability sets of entities. Our experiments show that our method finds biologically and statistically significant alignments in the order of milliseconds.
AVAILABILITY: Our software and the source code in C programming language is available at http://bioinformatics.cise.ufl.edu/pal.html.
Journal Website
Conference Papers
Although the problem of aligning metabolic networks has been considered in the past, the running time and space complexity of these solutions has so far limited their use to moderately sized networks. In this paper, we address the problem of aligning two metabolic networks, particularly when both of them are too large to be dealt with using existing methods. We develop a generic framework that can be used with any existing method to significantly improve the scale of the networks that can be aligned in practical time. Our framework has three major phases, namely the compression phase, the alignment phase and the refinement phase. For the first phase, we develop an algorithm which transforms the given networks to a compressed domain where they are summarized using fewer nodes, termed supernodes, and interactions. In the second phase, we carry out the alignment in the compressed domain using an existing method as our base algorithm. This alignment results in supernode mappings in the compressed domain, each of which are smaller instances of network alignment. In the third phase, we solve each of the instances using the base alignment algorithm
to refine the alignment results. Our experiments demonstrate that this method can reduce the sizes of metabolic networks by almost half at each compression level. For the overall framework, we demonstrate how well it increases the performance of an existing alignment method. We observe that we can align twice or more as large networks using the same amount of resources with our framework compared to a recent method for network alignment, namely SubMAP. Our results also suggest that the alignment obtained by only one level of compression captures the original alignment results with very high accuracy.
PDF
We consider the problem of aligning two metabolic pathways. Unlike traditional approaches, we do not restrict the alignment to one-to-one mappings between the molecules of the input pathways. We follow the observation that in nature different organisms can perform the same or similar functions through different sets of reactions and molecules. The number and the topology of the molecules in these alternative sets often vary from one organism to another. In other words, given two metabolic pathways of arbitrary topology, we would like to find a mapping that maximizes the similarity between the molecule subsets of query pathways of size at most a given integer k. We transform this problem into an eigenvalue problem. The solution to this eigenvalue problem produces alternative mappings in the form of a weighted bipartite graph. We then convert this graph to a vertex weighted graph. The maximum weight independent subset of this new graph is the alignment that maximizes the alignment score while ensuring consistency. We call our algorithm SubMAP (Subnetwork Mappings in Alignment of Pathways). We evaluate its accuracy and performance on real datasets. Our experiments demonstrate that SubMAP can identify biologically relevant mappings that are missed by traditional alignment methods and it is scalable for real size metabolic pathways.
PDF
Identifying steady states that characterize the long term outcome of regulatory networks is crucial in understanding important biological processes such as cellular differentiation. Finding all possible steady states of regulatory networks is a computationally intensive task as it suffers from state space explosion problem. Here, we propose a method for finding
steady states of large-scale Boolean regulatory networks. Our method exploits scale-freeness and weak connectivity of regulatory networks in order to speed up the steady state search through partitioning. In the trivial case where network has more than one component such that the components are disconnected from each other, steady states of each component are independent of those of the remaining components. When the size of at least one connected component of the network is still prohibitively large, further partitioning is necessary. In this case, we identify weakly dependent components (i.e., two components that have a small number of regulations from one to the other) and calculate the steady states of each such component independently. We then combine these steady states by taking into account the regulations connecting them. We show that this approach is much more efficient than calculating the steady states of the whole network at once when the number of edges connecting them is small. Since regulatory networks often have small in-degrees, this partitioning strategy can be used effectively in order to find their steady states. Our experimental results on real datasets demonstrate that our method leverages steady state identification to very large regulatory networks.
PDF
Analyzing metabolic pathways by means of their steady states has
proven to be accurate and efficient for practical purposes. The models
such as elementary flux modes (EFMs) and extreme pathways
(EPs) define the boundaries of the metabolic flux cone that is the
set of all steady states of a pathway. However, the contributions
of the subsets of pathway components in this flux cone so far has
not been characterized mathematically. Also, the functional similarities
of different component sets (e.g., sets of reactions) has not
been expressed as a function of the steady states of metabolic pathways.
Here, we aim to fill this gap by proposing a model that quantifies
the impact of a set of components on the steady states of a
pathway by using EFMs. At a high level, we model the impact of
a given component set as the change in the flux cone when all the
elements of that set are inhibited. Furthermore, given two sets of
components from different pathways, we measure their functional
similarity as the similarity of their impacts on corresponding pathways.
Computing this functional similarity is a computationally
challenging task as it requires finding the volumes of the intersection
and the union of two polyhedral cones in high dimensional
space. These volumes cannot be expressed in closed form. In this
paper, we develop a novel method that first transforms the polyhedral
cones to polytopes and then uses minimum enclosing balls to
approximate this intersection efficiently. Our experiments on real
metabolic pathways demonstrate that our method is of great use for
both measuring the impacts of pathway components and identifying
functionally similar component sets.
PDF
Often groups of genes in regulatory networks, also called modules, work collaboratively on similar functions. Mathematically, the modules in a regulatory network has often been thought as a group of genes that interact with each other significantly more than the rest of the network. Finding such modules is one of the fundamental problems in understanding gene regulation. In this paper, we develop a new approach to identify modules of genes with similar functions in biological regulatory networks (BRNs). Unlike existing methods, our method recognizes that there are different types of interactions (activation, inhibition), these interactions have directions and they take place only if the activity levels of the activating (or inhibiting) genes are above certain thresholds. Furthermore, it also considers that as a result of these interactions, the activity levels of the genes change over time even in the absence of external perturbations. Here we addresses both the dynamic behavior of gene activity levels and the different interaction types by an incremental algorithm that is scalable to the organism wide BRNs with many dynamic steps. Our experimental results suggest that our method can identify biologically meaningful modules that are missed by traditional approaches.
PDF
Background
Computing the long term behavior of regulatory and signaling networks is critical in understanding how biological functions take place in organisms. Steady states of these networks determine the activity levels of individual entities in the long run. Identifying all the steady states of these networks is difficult due to the state space explosion problem.
Methodology
In this paper, we propose a method for identifying all the steady states of Boolean regulatory and signaling networks accurately and efficiently. We build a mathematical model that allows pruning a large portion of the state space quickly without causing any false dismissals. For the remaining state space, which is typically very small compared to the whole state space, we develop a randomized traversal method that extracts the steady states. We estimate the number of steady states, and the expected behavior of individual genes and gene pairs in steady states in an online fashion. Also, we formulate a stopping criterion that terminates the traversal as soon as user supplied percentage of the results are returned with high confidence.
Conclusions
This method identifies the observed steady states of boolean biological networks computationally. Our algorithm successfully reported the G1 phases of both budding and fission yeast cell cycles. Besides, the experiments suggest that this method is useful in identifying co-expressed genes as well. By analyzing the steady state profile of Hedgehog network, we were able to find the highly co-expressed gene pair GL1-SMO together with other such pairs.
Availability
Source code of this work is available at http://bioinformatics.cise.ufl.edu/palSteady.html
PDF
Computing the long term behavior of regulatory and signaling networks is critical in understanding how biological functions take place in organisms. Steady states of these networks determine the activity levels of individual entities in the long run. Identifying all the steady states of these networks is difficult due to the state space explosion problem.
Methodology
In this paper, we propose a method for identifying all the steady states of Boolean regulatory and signaling networks accurately and efficiently. We build a mathematical model that allows pruning a large portion of the state space quickly without causing any false dismissals. For the remaining state space, which is typically very small compared to the whole state space, we develop a randomized traversal method that extracts the steady states. We estimate the number of steady states, and the expected behavior of individual genes and gene pairs in steady states in an online fashion. Also, we formulate a stopping criterion that terminates the traversal as soon as user supplied percentage of the results are returned with high confidence.
Conclusions
This method identifies the observed steady states of boolean biological networks computationally. Our algorithm successfully reported the G1 phases of both budding and fission yeast cell cycles. Besides, the experiments suggest that this method is useful in identifying co-expressed genes as well. By analyzing the steady state profile of Hedgehog network, we were able to find the highly co-expressed gene pair GL1-SMO together with other such pairs.
Availability
Source code of this work is available at http://bioinformatics.cise.ufl.edu/palSteady.html
Pathways show how different biochemical entities interact with each other to perform vital functions for the survival
of organisms. Similarities between pathways indicate functional similarities that are difficult to identify by comparing
the individual entities that make up those pathways. When interacting entities are of single type, the problem of
identifying similarities reduces to graph isomorphism problem. However, for pathways with varying types of entities,
such as metabolic pathways, alignment problem is more challenging. Existing methods, often, address the metabolic
pathway alignment problem by ignoring all the entities except for one type. This kind of abstraction reduces the
relevance of the alignment significantly as it causes losses in the information content. In this paper, we develop a
method to solve the pairwise alignment problem for metabolic pathways. One distinguishing feature of our method
is that it aligns reactions, compounds and enzymes without abstraction of pathways. We pursue the intuition that
both pairwise similarities of entities (homology) and their organization (topology) are crucial for metabolic pathway
alignment. In our algorithm, we account for both by creating an eigenvalue problem for each entity type. We enforce
the consistency by considering the reachability sets of the aligned entities. Our experiments show that, our method
finds biologically and statistically significant alignments in the order of seconds for pathways with ∼ 100 entities.
PDF